You’re holding a deep purple amethyst bracelet under a lamp, and it suddenly looks richer—almost electric. In the studio, I’ve had that exact moment while sorting gemstones for StarryBead designs, and it always triggers the same question: why do some materials “catch” light and color so differently? Chemistry has a practical answer for many colored compounds: crystal field theory. It explains how a metal ion’s electrons respond when surrounded by nearby atoms or molecules, and how that can change color and magnetism in a predictable way.
Crystal field theory is one of those topics that sounds intimidating, but it’s really a story about crowding. When ligands (the neighbors around a metal ion) get close, they push on the metal’s d orbitals, splitting them into groups of different energy. That splitting controls what wavelengths of light are absorbed—and that’s where color often comes from.

What Is Crystal Field Theory (CFT) in Plain English?
Crystal field theory (CFT) models the interaction between a central metal ion and surrounding ligands as mostly electrostatic—like charges and dipoles pushing and pulling. It’s not the full bonding picture (that’s where ligand field theory and MO theory go further), but CFT is fast, visual, and surprisingly powerful for explaining trends.
Here’s the core idea:
- A free metal ion has five d orbitals at the same energy (degenerate).
- Surround it with ligands, and repulsion is not equal for every orbital direction.
- So the d orbitals split into higher- and lower-energy sets.
That’s crystal field theory in one breath: ligands split d-orbital energies.
The Big Concept: Why d Orbitals Split
The five d orbitals point in different directions:
- d(x²−y²) and d(z²) aim along axes (they “stick out” where ligands often sit).
- d(xy), d(xz), d(yz) point between axes.
If ligands sit on axes, orbitals pointing at them feel more repulsion and become higher energy. Orbitals pointing between ligands feel less repulsion and become lower energy. That energy gap is the crystal field splitting energy (often written Δ).
This is why crystal field theory connects directly to properties you can measure:
- Color (which light gets absorbed)
- Magnetism (how many electrons stay unpaired)
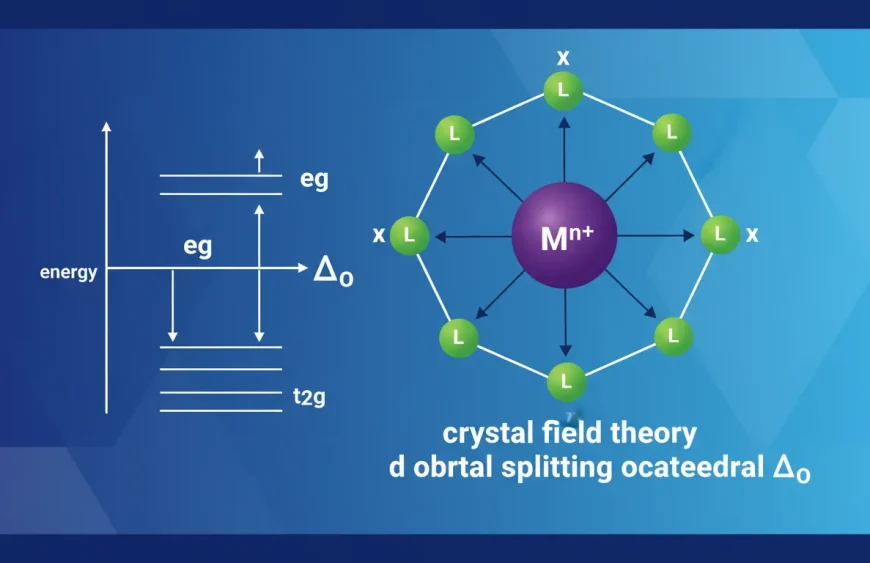
Octahedral Splitting (The Most Common Diagram)
In an octahedral complex, six ligands surround the metal (like points on a 3D cross). Two d orbitals get pushed up, three drop down:
- Higher energy: eg = d(x²−y²), d(z²)
- Lower energy: t2g = d(xy), d(xz), d(yz)
- Splitting size: Δo
Why it matters: If Δo is large enough, electrons prefer pairing in the lower set. If Δo is small, electrons spread out to avoid pairing. That single tradeoff drives “high spin vs low spin.”
High Spin vs Low Spin: The Pairing vs Promotion Decision
In crystal field theory, electrons choose between:
- Pairing energy (P): energy cost to pair two electrons in one orbital
- Promotion across Δ: energy cost to place an electron in a higher set
So:
- Weak-field ligands → smaller Δo → high spin (more unpaired electrons)
- Strong-field ligands → larger Δo → low spin (fewer unpaired electrons)
A practical memory hook (not perfect, but useful): ligands like CN⁻ and CO tend to be strong-field; halides like Cl⁻ tend to be weak-field. For a deeper reference overview, see Crystal field theory (Wikipedia) and the more textbook-style walkthrough at Chem LibreTexts: Crystal Field Theory.
| Geometry | Splitting pattern | Typical Δ size (relative) | High spin common? | Typical magnetic behavior | Color trend (vs Δ) | Example ions (common cases) |
|---|---|---|---|---|---|---|
| Octahedral | t2g (lower) / eg (higher) | Medium (baseline) | Yes (esp. 3d with weak-field ligands) | Often more paramagnetic (HS); can be less if LS | Moderate; larger Δ often gives weaker/longer-λ absorption | Fe²⁺ (HS common), Co³⁺ (LS common with strong-field) |
| Tetrahedral | e (lower) / t2 (higher) | Small (≈4/9 of octahedral) | Yes | More paramagnetic (unpaired electrons typical) | Often stronger (smaller Δ → visible-range d–d; intense also due to less strict selection rules) | Fe²⁺ (HS), Ni²⁺ (often Td in some salts) |
| Square planar | (dsp²) typically: dx²−y² highest; dxy next; dz²; dxz/dyz lowest | Large (for many d⁸ metals) | No (often low spin) | Less paramagnetic (often diamagnetic for d⁸) | Often weaker/shifted (large Δ can push absorption out of visible) | Ni²⁺ (d⁸, often square planar), Cu²⁺ (often square planar/distorted) |
Tetrahedral vs Octahedral: Same Idea, Different Split
In tetrahedral complexes (four ligands), the splitting pattern flips relative to octahedral naming and is usually smaller:
- Lower energy: e (two orbitals)
- Higher energy: t2 (three orbitals)
- Splitting size: Δt, typically ~4/9 of Δo (rule of thumb)
Because Δt is smaller, tetrahedral complexes are usually high spin—there’s just not enough splitting to force extensive pairing.
Square Planar: Why Some Complexes Go Flat
Some d⁸ metals (especially Ni²⁺, Pd²⁺, Pt²⁺) form square planar complexes with a very large splitting pattern that strongly favors pairing. Crystal field theory helps you anticipate:
- Low spin behavior
- Often diamagnetism (no unpaired electrons) in classic square planar d⁸ cases
This is one place where CFT diagrams become a “property prediction tool,” not just a drawing exercise.
How Crystal Field Theory Explains Color (What You Actually See)
Color often comes from d–d transitions:
- Light hits a complex.
- An electron absorbs a photon with energy matching Δ.
- The electron “jumps” from lower d set to higher d set.
- The remaining transmitted/reflected light looks colored (complementary color).
So, bigger Δ → higher-energy (shorter wavelength) absorption. Smaller Δ → lower-energy (longer wavelength) absorption. That’s why changing the ligand, oxidation state, or geometry can shift observed color.
If you want a clean, beginner-friendly explanation with diagrams and practice-style framing, Chad’s Prep crystal field theory lesson is a solid companion.

How Crystal Field Theory Explains Magnetism (Why Some Compounds Act Like Tiny Magnets)
Magnetism, in many coordination compounds, mostly tracks unpaired electrons:
- More unpaired electrons → more paramagnetic
- All paired → diamagnetic
Crystal field theory tells you whether electrons will pair up (low spin) or stay unpaired (high spin) by comparing Δ and pairing energy. That’s why the same metal can show different magnetism depending on ligands and geometry.
A quick, usable checklist:
- Determine metal oxidation state → d-electron count.
- Identify geometry (octahedral, tetrahedral, square planar).
- Estimate ligand strength → predict Δ size.
- Fill split orbitals → count unpaired electrons → predict magnetic behavior.
A Short Story From the Bench: Where People Get Confused (And How I Unstick It)
When I first learned crystal field theory, I kept mixing up “which orbitals go up” and “why color changes.” What fixed it for me wasn’t memorizing diagrams—it was picturing ligands as streetlights on the axes. If an orbital points toward the streetlights, it gets “glared at” (repelled) and rises in energy. Once that clicked, the rest (Δ, color, high/low spin, magnetism) became one connected chain instead of separate facts.
If you’re learning this for a class, I also recommend drawing the axes every time. It slows you down in a good way—and prevents the classic eg/t2g flip error.
Crystal Field Theory: Strengths and Limits (Quick, Honest Take)
Crystal field theory works best when you need trends and predictions:
- Geometry → splitting pattern
- Ligand strength → Δ size trend
- Δ vs pairing → spin state
- Spin state → magnetism
- Δ → absorption → color shifts
But CFT can fall short because it treats bonding as mostly electrostatic. Real bonding can include covalency and π effects, which is why more advanced treatments (ligand field theory / MO theory) sometimes become necessary—especially for detailed spectra.
Connecting the Chemistry Back to StarryBead (Without Overclaiming)
StarryBead focuses on natural crystals and gemstones for wellness and style—those materials get their colors from a variety of causes (trace metals, defects, charge transfer, and more). Crystal field theory is one of the key chemistry tools that helps explain why transition-metal-containing materials can show vivid, tunable colors. It’s not the whole gemstone-color story, but it’s a powerful piece of the puzzle—and it’s a great reminder that beauty often has a blueprint.
If you enjoy learning the science behind color and materials, you’ll likely also love browsing pieces that highlight natural variation—because that variation often starts at the atomic level.
Conclusion: The Simple Takeaway You Can Keep
Crystal field theory is the chemistry of “neighbors changing your energy.” Ligands crowd a metal ion, split its d orbitals, and that splitting helps explain colors and magnetic behavior in coordination compounds. The next time you notice a dramatic color shift under different lighting, you’ll have a new lens: somewhere in that material’s structure, electrons are responding to their local field.
FAQ: Crystal Field Theory Questions People Also Search
1) What does crystal field theory explain?
It explains how ligands split a metal ion’s d-orbital energies, helping predict color, magnetism, and high-spin/low-spin behavior.
2) Why do d orbitals split in an octahedral complex?
Because ligands approach along axes and repel orbitals pointing directly at them (eg) more than those pointing between axes (t2g).
3) What is Δo in crystal field theory?
Δo is the octahedral crystal field splitting energy—the energy gap between t2g and eg sets.
4) How does crystal field theory relate to color?
Color can come from d–d transitions where electrons absorb light with energy equal to Δ; the observed color is often complementary to the absorbed wavelength.
5) What’s the difference between high spin and low spin?
High spin means more unpaired electrons (small Δ); low spin means more paired electrons (large Δ), mainly in octahedral complexes with certain d counts.
6) Are tetrahedral complexes usually high spin?
Yes, because Δt is typically smaller, making pairing less favorable.
7) What are the limitations of crystal field theory?
It simplifies bonding as electrostatic and may not capture covalency/π bonding effects that influence real spectra and properties.











